💊 The FDA Is Unsafe and Ineffective
The 82X Cost Multiplier: Two Systems, Two Outcomes
The Oxford RECOVERY trial tested COVID treatments on 47,000 patients for $500 per patient.
The FDA-regulated system averages $41,000 per patient.
That’s 82X more expensive.
The RECOVERY trial found dexamethasone saves lives. Time from start to result: 3 months. FDA average time to approve a new drug: 9.1 years from Phase 1.
That’s 36X slower (9.1 years ≈ 109 months ÷ 3 months).
Combined inefficiency: 82 × 36 = 2,952X less efficient overall.
These aren’t estimates. These are documented outcomes from actual trials.
Historical Evidence: How We Broke Medicine
We’re only 2 lifetimes from the use of the modern scientific method in medicine. Thus, it’s only been applied for 0.0001% of human history. The more clinical research studies we read, the more we realize we don’t know. Nearly every study ends with the phrase “more research is needed”. We know basically nothing at this point compared to what will eventually be known about the human body.
There is compelling historical evidence suggesting that large scale efficacy-trials based on real-world evidence have ultimately led to better health outcomes than current pharmaceutical industry-driven randomized controlled trials.
For over 99% of recorded human history, the average human life expectancy has been around 30 years.
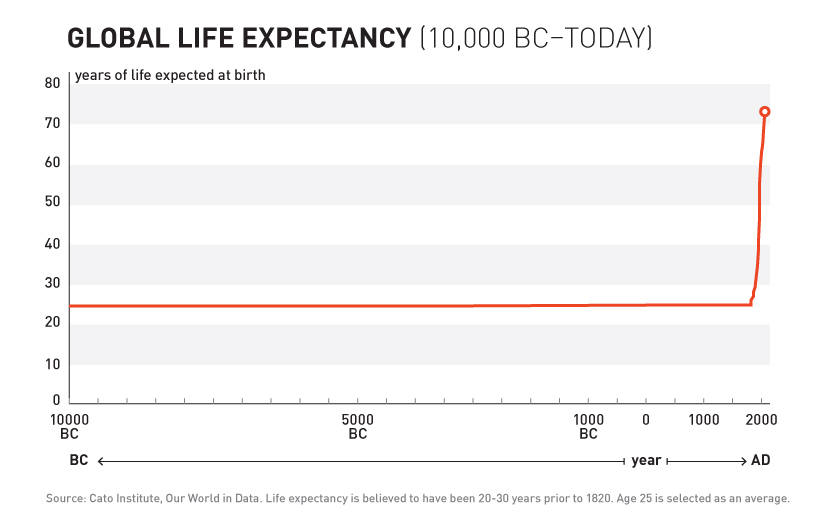
1893 - The Advent of Safety and Efficacy Trials
In the late nineteenth and early twentieth century, clinical objectivity grew. The independent peer-reviewed Journal of the American Medical Association (JAMA) was founded in 1893. It would gather case reports from the 144,000 physicians members of the AMA on the safety and effectiveness of drugs. The leading experts in the area of a specific medicine would review all of the data and compile them into a study listing side effects and the conditions for which a drug was or was not effective. If a medicine were found to be safe, JAMA would give its seal of approval for the conditions where it was found to be effective.
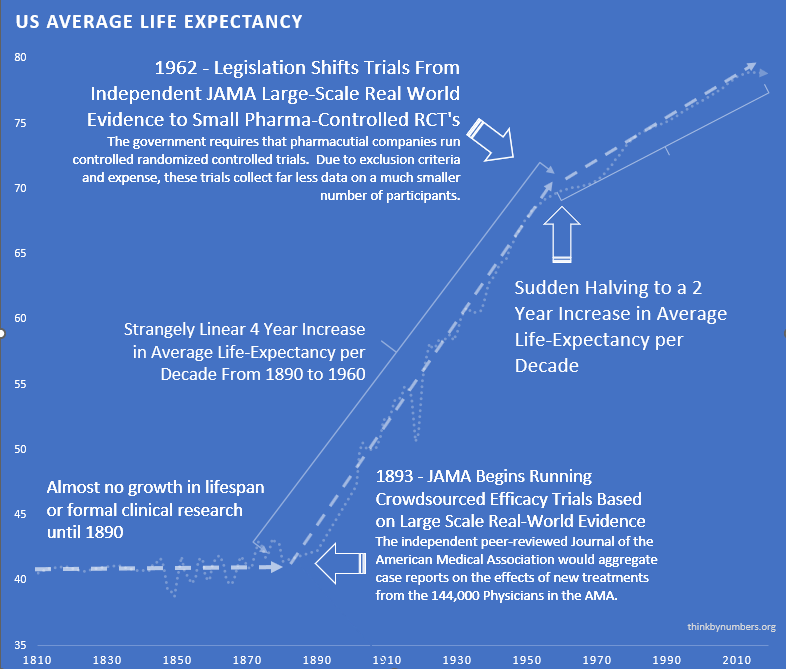
The adoption of this system of crowd-sourced, observational, objective, and peer-reviewed clinical research was followed by a sudden shift in the growth of human life expectancy. After over 10,000 years of almost no improvement, we suddenly saw a strangely linear 4-year increase in life expectancy every decade.
1938 - The FDA Requires Phase 1 Safety Trials
A drug called Elixir sulfanilamide caused over 100 deaths in the United States in 1937.
Congress reacted to the tragedy by requiring all new drugs to include:
“adequate tests by all methods reasonably applicable to show whether or not such drug is safe for use under the conditions prescribed, recommended, or suggested in the proposed labeling thereof.”
These requirements evolved to what is now called the Phase 1 Safety Trial.
This consistent four-year/year increase in life expectancy remained unchanged before and after the new safety regulations.
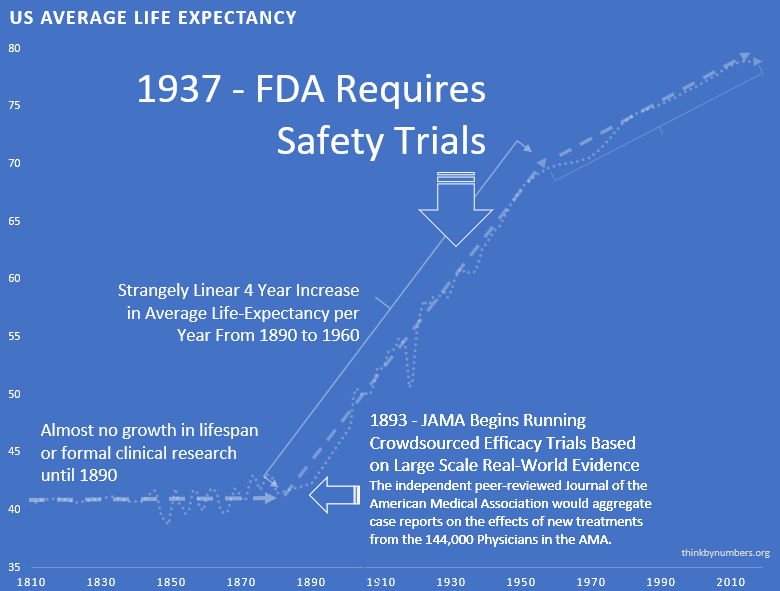
This suggests that the regulations did not have a large-scale positive or negative impact on the development of life-saving interventions.
1950’s - Thalidomide Causes Thousands of Birth Defects Outside US
Thalidomide was first marketed in Europe in 1957 for morning sickness. While it was initially thought to be safe in pregnancy, it resulted in thousands of horrific congenital disabilities.
Fortunately, the existing FDA safety regulations prevented any birth defects in the US. Despite the effectiveness of the existing US regulatory framework in protecting Americans, newspaper stories such as the one below created a strong public outcry for increased regulation.

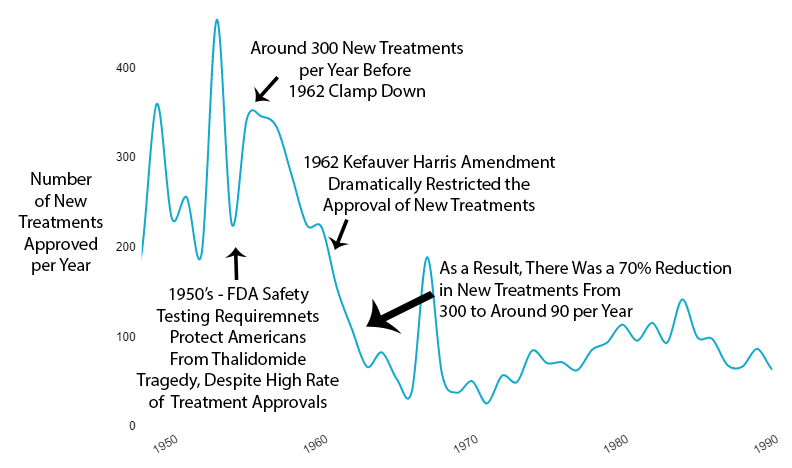
1962 - New Efficacy Regulations Reduce the Amount and Quality of Efficacy Data Collected
As effective safety regulations were already in place, the government instead responded to the Thalidomide disaster by regulating efficacy testing via the 1962 Kefauver Harris Amendment. Before the 1962 regulations, it cost a drug manufacturer an average of $74 million (2020 inflation-adjusted) to develop and test a new drug for safety before bringing it to market. Once the FDA had approved it as safe, efficacy testing was performed by the third-party American Medical Association. Following the regulation, trials were instead to be conducted in small, highly-controlled trials by the pharmaceutical industry.
Reduction in Efficacy Data
The 1962 regulations made these large real-world efficacy trials illegal. Ironically, even though the new regulations were primarily focused on ensuring that drugs were effective through controlled FDA efficacy trials, they massively reduced the quantity and quality of the efficacy data that was collected for several reasons:
- New Trials Were Much Smaller
- Participants Were Less Representative of Actual Patients
- They Were Run by Drug Companies with Conflicts of Interest Instead of the 3rd Party AMA
Reduction in New Treatments
The new regulatory clampdown on approvals immediately reduced the production of new treatments by 70%.
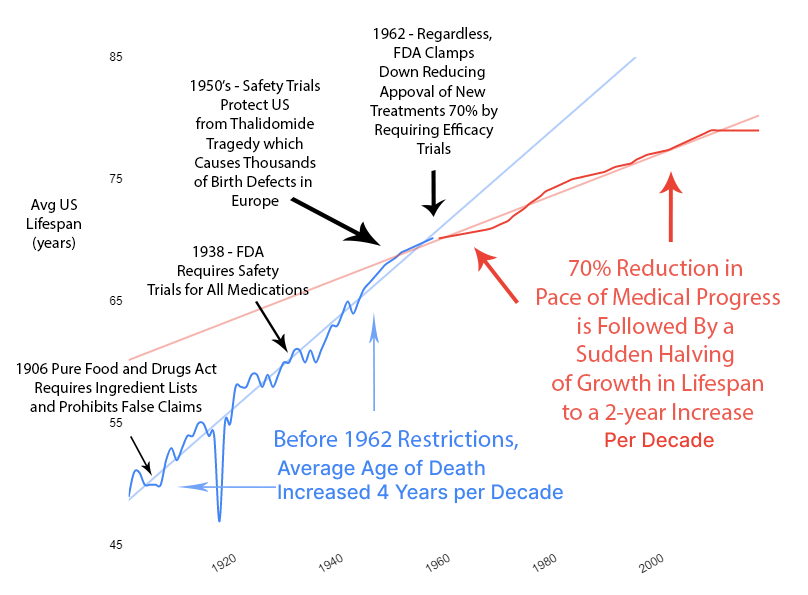
Reduction in Life Expectancy Gains
The impact on human life expectancy was immediate and devastating. Before 1962, life expectancy increased by 4 years every decade. After 1962, that rate was cut in half to just 2 years per decade.
Explosion in Costs
Since the abandonment of the former efficacy trial model, costs have exploded. Since 1962, the cost of bringing a new treatment to market has gone from $74 million to over $1 billion US dollars (2020 inflation-adjusted).
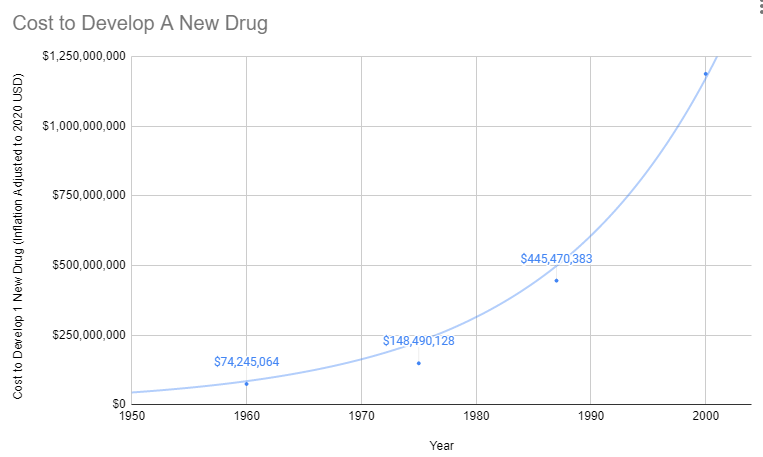
The Correlation: Rising Costs, Stagnating Lifespans
As development costs skyrocketed, life expectancy gains flatlined. The correlation is undeniable:
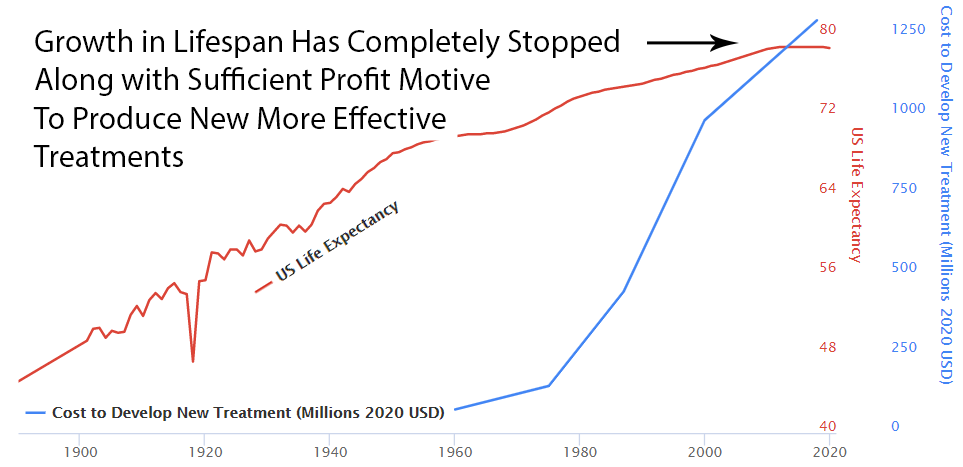
High Cost of Development Favors Monopoly and Punishes Innovation
There’s another problem with the increasing costs of treatment development. In the past, a genius scientist could come up with a treatment, raise a few million dollars, and do safety testing. Now that it costs a billion dollars to get a drug to market, the scientist has to persuade one of a few giant drug companies that can afford it to buy his patent.
Then the drug company has two options:
Option 1: Risk $1 billion on clinical trials
Possibility A: Drug turns out to be one of the 90% the FDA rejects. GIVE BANKER A BILLION DOLLARS. DO NOT PASS GO.
Possibility B: Drug turns out to be one of the 10%, the FDA approves. Now it’s time to try to recover that billion dollars. However, very few drug companies have enough money to survive this game. So, this company almost certainly already has an existing inferior drug on the market to treat the same condition. Hence, any profit they make from this drug will likely be subtracted from revenue from other drugs they’ve already spent a billion dollars on.
Option 2: Put the patent on the shelf
Do not take a 90% chance of wasting a billion dollars on failed trials. Do not risk making your already approved cash-cow drugs obsolete.
What’s the benefit of bringing better treatment to market if you’re just going to lose a billion dollars? Either way, the profit incentive is entirely in favor of just buying better treatments and shelving them.
Cures Are Far Less Profitable Than Lifetime Treatments
If the new treatment is a permanent cure for the disease, replacing a lifetime of refills with a one-time purchase would be economically disastrous for the drug developer. With a lifetime prescription, a company can recover its costs over time. Depending on the number of people with the disease, one-time cures would require a massive upfront payment to recover development costs.
How is there any financial incentive for medical progress at all?
Fortunately, there isn’t a complete monopoly on treatment development. However, the more expensive it is to get a drug to market, the fewer companies can afford the upfront R&D investment. So the drug industry inevitably becomes more monopolistic. Thus there are more situations where the cost of trials for a superior treatment exceeds the profits from existing treatments.
Lack of Incentive to Discover New Applications for Off-Patent Treatments
There are roughly 10,000 known diseases afflicting humans, most of which (approximately 95%) are classified as “orphan” (rare) diseases. The current system requires that a pharmaceutical company predict a particular condition in advance of running clinical trials. If a drug is found to be effective for other diseases after the patent has expired, no one has the financial incentive to get it approved for another disease.
People With Rare Disease are Severely Punished
In the case of rare diseases, increasing the cost of treatment development to over a billion makes it impossible to recover your investment from a small number of patients. So rare disease patients suffer the most severe harm from the added regulatory burden on development.
How high should the cost of drug development be on our list of human problems? Well, when something costs more, you get less of it. For people dying of cancer, the fact that we couldn’t afford enough research to cure them is definitely at the top of their list of human problems.
Delayed Life-Saving Treatments
One unanticipated consequence of the amendments was that the new burden of proof made the process of drug development both more expensive and much longer, leading to increasing drug prices and a “drug lag”. After that point, whenever they released some new cancer or heart medication that would save 50 thousand lives a year, it meant that over the previous ten years of trials, 500,000 people died because they didn’t have access to the drug earlier.
Deaths Due to US Regulatory “Drug Lag”
A comparative analysis between countries suggests that delays in new interventions cost anywhere from 21,000 to 120, 000 US lives per decade.
Deaths owing to drug lag have been numbered in the hundreds of thousands. It’s estimated that practolol, a drug in the beta-blocking family, could save ten thousand lives a year if allowed in the United States. Although the FDA allowed a first beta-blocker, propranolol, in 1968, three years after that drug had been available in Europe, it waited until 1978 to allow propranolol to treat hypertension and angina pectoris, its most essential indications. Despite clinical evidence as early as 1974, only in 1981 did the FDA allow a second beta-blocker, timolol, to prevent a second heart attack. The agency’s withholding of beta-blockers was alone responsible for probably tens of thousands of deaths.
Data from the Tufts Center for the Study of Drug Development suggests that thousands of patients have died because of US regulatory delays relative to other countries, for new drugs and devices, including:
- interleukin-2
- Taxotere
- vasoseal
- ancrod
- Glucophage
- navelbine
- Lamictal
- ethyol
- photofrin
- rilutek
- citicoline
- panorex
- Femara
- ProStar
- omnicath
Before US FDA approval, most of these drugs and devices had already been available in other countries for a year or longer.
Following the 1962 increase in US regulations, one can see a divergence from Switzerland’s growth in life expectancy, which did not introduce the same delays to availability.
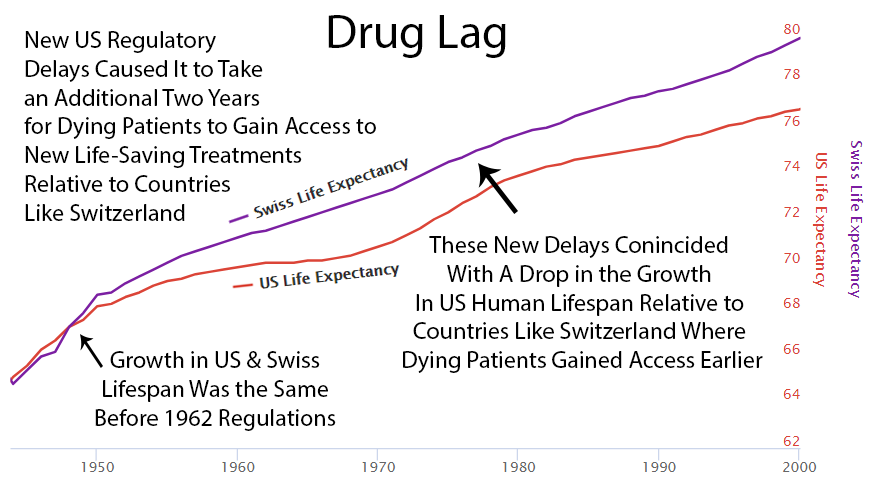
Perhaps it’s a coincidence, but you can see an increase in drug approvals in the ’80s. At the same time, the gap between Switzerland and the US gets smaller. Then US approvals go back down in the ’90s, and the gap expands again.
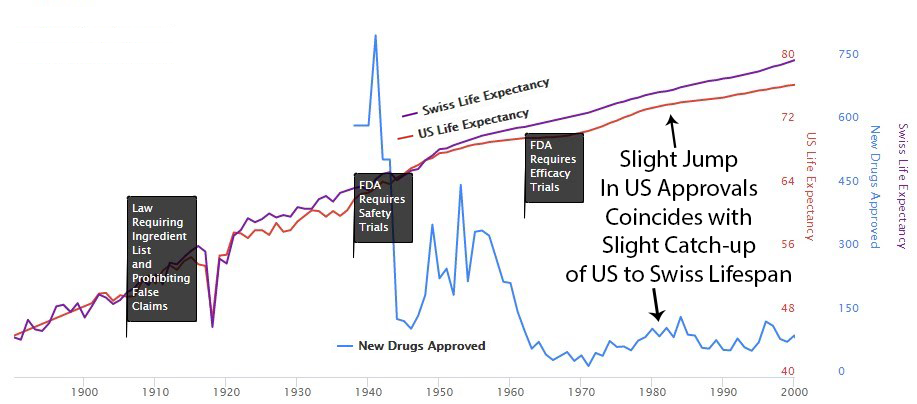
Increase in Patent Monopoly
Industry agitation surrounding the “drug lag” finally led to the modification of the drug patenting system in the Drug Price Competition and Patent Term Restoration Act of 1984. This further extended the life of drug patents. Thus Kefauver’s amendments ultimately made drugs more expensive by granting longer monopolies.
Decreased Ability to Determine Comparative Efficacy
The placebo-controlled, randomized controlled trial helped researchers gauge the efficacy of an individual drug. However, it makes the determination of comparative effectiveness much more difficult.
Slowed Growth in Life Expectancy
From 1890 to 1960, there was a linear 4-year increase in human lifespan every decade. This amazingly linear growth rate had followed millennia with a flat human lifespan of around 28 years. Following this new 70% reduction in the pace of medical progress, the growth in human lifespan was immediately cut in half to an increase of 2 years per decade.
Progress! The FDA’s current system works like this:
- Scientists discover a cure for your disease
- You wait 17 years
- You die
- The cure gets approved
- Pharmaceutical companies charge your widow $10,000 per pill
It’s a beautiful system if you’re a mortician or a bankruptcy lawyer.
Addressing Common Objections
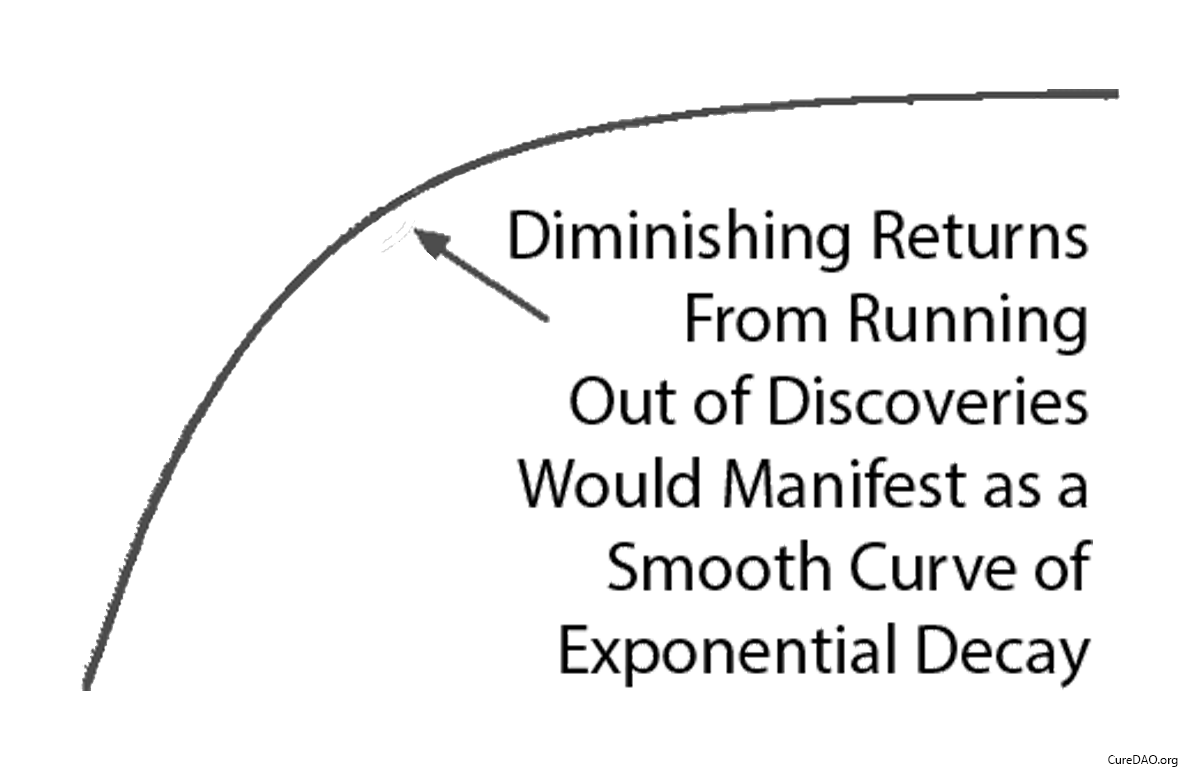
Diminishing Returns?
One might say “It seems more likely — or as likely — to me that drug development provides diminishing returns to life expectancy.” However, diminishing returns produce a slope of exponential decay. It may be partially responsible, but it’s not going to produce a sudden change in the linear slope of a curve a linear as life expectancy was before and after the 1962 regulations.
Correlation is Not Causation
You might say “I don’t know how much the efficacy regulations contribute to or hampers public health. I do know that correlation does not necessarily imply causation.” However, a correlation plus a logical mechanism of action is the least bad method we have for inferring the most likely significant causal factor for an outcome (i.e. life expectancy). Assuming most likely causality based on temporal correlation is the entire basis of a clinical research study and the scientific method generally.
Impact of Innovative Medicines on Life Expectancy
A three-way fixed-effects analysis of 66 diseases in 27 countries, suggests that if no new drugs had been launched after 1981, the number of years of life lost would have been 2.16 times higher it actually was. It estimates that pharmaceutical expenditure per life-year saved was $2837.

More people survive as more treatments are developed. There’s a strong correlation between the development of new cancer treatments and cancer survival over 30 years.

The $2.6 Billion Price Tag for Aspirin
Here’s what it actually costs to get FDA approval for a new drug:
- Average cost: $2.6 billion
- Average time: 9.1 years from Phase 1 (17 years total discovery-to-patient)
- Success rate: 12% make it through trials
That’s $41,000 per person just to TEST if a pill works. Not the pill. Just to test it.
For that money, you could: - Buy a Tesla and crash it into the disease - Hire a shaman for a year - Fly to Switzerland for assisted dying (much faster)
The total cost? $2.6 billion per new drug.
That’s more than the GDP of Greenland. We could literally buy Greenland and turn it into a giant laboratory for less than it costs to approve one arthritis medication.
Fun fact: If we had today’s FDA in 1897, aspirin would still be in Phase II trials. “We need to test if it works on headaches in left-handed people born on Tuesdays.”
17 years from discovery to patient (while you die waiting).
That’s: - 4 presidential terms - 2 new iPhone models becoming obsolete - Your entire child growing up and moving out - You dying of whatever the cure was for
But at least the paperwork is thorough!
95% of diseases have ZERO approved treatments.
Your rare disease child has better odds with lottery tickets.
Why? Math. If your disease only affects 10,000 people, and it costs $2.6 billion to develop a drug, each patient would need to pay $260,000 just to break even.
So pharmaceutical companies focus on: - Erectile dysfunction (affects: half of all men’s egos) - Baldness (affects: men with money) - Wrinkles (affects: women with money) - Cholesterol (affects: everyone who eats)
Meanwhile, if you have Exploding Kidney Syndrome affecting 37 people worldwide, the FDA’s message is: “Have you tried not having Exploding Kidney Syndrome?”
The 166 Billion Treatments We’re Ignoring
There are 166 billion potential drug compounds we could test.
Number we’ve actually tested thoroughly: About 2,000.
That’s like having a library with 166 billion books but only reading the ones with pictures.
At our current rate of testing, we’ll have evaluated all possible treatments by the year 3,847,293.
Hope your descendants enjoy the cure for cancer!
Designed to prevent lawsuits, not save lives.
No Data on Unpatentable Molecules
Under the current system of research, it costs $41k per subject in Phase III clinical trials. As a result, there is not a sufficient profit incentive for anyone to research the effects of any factor besides a molecule that can be patented.

The Actual Death Toll (With Real Math)
The FDA’s own economists have calculated the body count from these delays. They call it “drug lag,” which is a sterile, bureaucratic term for “people dying while we shuffle papers.” The cost is staggering: delays in new treatments are estimated to cost anywhere from 21,000 to 120,000 American lives per decade.
A landmark case is beta-blockers. While Europe was happily using them to prevent heart attacks, the FDA waited. And waited. Propranolol was available in Europe three years before the US, but even after finally letting it in the door in 1968, the agency dithered. It waited until 1978—a full decade—to approve it for its most essential use, hypertension. Then it waited until 1981—a full sixteen years after it was available elsewhere—to approve a second beta-blocker to prevent a second heart attack.
The cost of this caution? - Beta-blockers delay: Gieringer (1985) estimated 10,000+ excess deaths per year during the decade-long delay. - Total beta-blocker deaths: ~100,000 Americans.
That’s more Americans than died in Vietnam and Korea combined. From one drug delay. And it wasn’t a one-off tragedy. Here is a partial list of other drugs Americans died waiting for while they were already available in other civilized countries for a year or longer:
We’re at the very beginning of thousands or millions of years of systematic discovery. So it’s unlikely that this decline in lifespan growth is the result of diminishing returns due to our running out of things to discover.
However, to validate the theory that large-scale real-world evidence can produce better health outcomes requires further validation of this method of experimentation. That’s the purpose of deFDA.
- interleukin-2
- Taxotere
- vasoseal
- ancrod
- Glucophage
- navelbine
- Lamictal
- ethyol
- photofrin
- rilutek
- citicoline
- panorex
- Femara
- ProStar
- omnicath
Each name on that list represents a graveyard of preventable deaths.
Case Study: The Human Cost of Regulatory Delay in Closed-Loop Neuromodulation
The historical data is damning, but this isn’t a problem of the past. Here is a modern example of the ongoing human cost of regulatory delay, focusing on a breakthrough technology for treatment-resistant depression.
Executive Summary
Depression is the leading global cause of disability and the strongest behavioral predictor of suicide. Existing antidepressant drugs fail to achieve remission in about two-thirds of patients, and pharmacologic innovation has stagnated. Closed-loop neuromodulation—implantable or wearable systems that sense neural activity and adjust stimulation in real time—has shown response rates of 60–80 % in pilot trials for treatment-resistant depression and obsessive–compulsive disorder. Yet under current FDA and EMA processes, large-scale availability is unlikely before 2035. A utilitarian cost-benefit model indicates that this delay will cost ≈5–10 million lives and ≈100 million quality-adjusted life-years (QALYs) globally.
1. Scale of the Problem
| Metric | Value | Source |
|---|---|---|
| Global major-depressive-disorder prevalence | ~280 million people | WHO 2023 |
| Annual suicides | ~700 000 | WHO 2023 |
| Suicide attributable to untreated or treatment-resistant depression | 70 % | CDC, WHO meta-analyses |
| Mean age of death | 40 y | Global Burden of Disease |
| Average lost QALYs per suicide | 35 | conservative midlife estimate |
Annual QALY loss:
700 000 × 35 = 24.5 million QALYs directly, plus ~2× from comorbid mortality ⇒ ≈75 million QALYs/year.
2. Expected Benefit of Closed-Loop Neuromodulation
Clinical prototypes (Stanford, UCSF, NIH):
- Acute response: 60–80 % of TRD patients improve ≥50 % on MADRS.
- Remission maintenance at 12 mo: 45–60 %.
- Adverse-event rate < 1 % serious, none fatal.
If global deployment reached 20 % of resistant cases (≈50 million candidates) with 50 % success, that yields:
- 25 million major depressive remissions.
- Suicide reduction ≈ 35 %.
- Indirect disease-mortality reduction ≈ 20 %.
Resulting life-saving potential: 0.35 × 700 000 × 10 = 2.45 million suicides averted per decade, plus ~5 million prevented indirect deaths.
3. Economic and Utility Calculation
| Variable | Estimate |
|---|---|
| QALYs gained per patient per year of remission | 0.5–0.8 |
| Patients benefited | 25 million |
| Annual QALY gain | ~15 million |
| Monetary valuation (US $100 000/QALY) | $1.5 trillion/year social value |
Each year of regulatory delay thus destroys ≈$1.5 trillion in global welfare—greater than the annual R&D budget of the entire pharmaceutical industry.
4. Comparative Timelines
| Scenario | Time to broad release | Cumulative preventable deaths (10 y horizon) |
|---|---|---|
| Current FDA/EMA process | 10–12 y | 5–10 million |
| Utilitarian adaptive approval | 2–3 y | 1–2 million |
| Net lives saved by reform | — | ≈8 million |
5. Feasibility of Accelerated Path
Key enablers of 24–36 month rollout:
- Adaptive Bayesian trials: Continuous learning eliminates phase segmentation.
- Dynamic e-consent: Allows real-time participation expansion.
- Automated adverse-event monitoring: Reduces manual inspection delays.
- Outcome-based conditional reimbursement: Pay per sustained remission.
With modern data infrastructure, these systems are already technically viable.
6. Conclusion
A decade of regulatory delay in closed-loop neuromodulation equates to roughly one genocide-scale loss of life and trillions of dollars in foregone human utility. From a utilitarian calculus, any risk threshold that delays access longer than 2–3 years imposes a harm orders of magnitude greater than potential device side-effects. A rational health-policy framework would authorize conditional deployment once expected QALYs gained exceed expected QALYs lost—a threshold crossed today.
The Incentive Structure: A Masterpiece of Misalignment
FDA Regulator Decision Tree:
Approve drug that later shows problems:
- Congressional hearing (televised)
- “FDA APPROVED KILLER DRUG” headlines
- Career ends
- Pension threatened
Delay drug that could save lives:
- Nothing happens
- Dead people don’t complain
- Promotion on schedule
- Retire to pharma job
This isn’t conspiracy. It’s economics. The FDA literally cannot make mistakes of commission (approving bad drugs) but faces zero consequences for mistakes of omission (delaying good drugs).
Clinical Trial Theater: Excluding 85% of Reality
FDA trials systematically exclude:
- Patients over 65 (most people who need drugs)
- Patients under 18 (all children)
- Pregnant women (50% of women sometimes)
- Anyone with comorbidities (everyone sick)
- Anyone on other medications (everyone old)
- Anyone who lives too far from a trial site (hope you’re rich and urban)
- Anyone who once looked at an aspirin
It’s like having a cure for cancer but only giving it to people who don’t have cancer.
Result: Test drugs on the 15% of unusually healthy sick people, then act shocked when they don’t work on actual patients.
Clinical trials have more exclusion criteria than a country club in 1950s Alabama. You must be exactly the right kind of sick - not too sick, not too healthy, and definitely not poor or old or pregnant or on any other medications.
Genius!
It’s like testing car safety using only crash test dummies, then being surprised when real humans with bones have different outcomes.
No Long-Term Outcome Data
Even if there is a financial incentive to research a new drug, there is no data on the long-term outcomes of the drug. The data collection period for participants can be as short as several months. Under the current system, it’s not financially feasible to collect data on a participant for years or decades. So we have no idea if the long-term effects of a drug are worse than the initial benefits.
For instance, even after controlling for co-morbidities, the Journal of American Medicine recently found that long-term use of Benadryl and other anticholinergic medications is associated with an increased risk for dementia and Alzheimer’s disease.
The FDA Requires Psychic Powers
Before you can test a drug, you must tell the FDA EXACTLY what it will cure and EXACTLY how you’ll measure that cure.
“This drug will reduce depression by 37.2% as measured by the Hamilton Rating Scale on Tuesdays.”
What if it cures depression by 90% but only on Wednesdays? Failed trial. What if it doesn’t cure depression but eliminates anxiety? Failed trial. What if it cures cancer instead? Failed trial, also please report to prison.
It’s like requiring Columbus to file a detailed map of America before letting him sail west.
In reality, this isn’t hypothetical. When running an efficacy trial, the FDA expects that the drug developer has the psychic ability to predict which conditions a treatment will be most effective for in advance of collecting the human trial data. If it was possible to magically determine this without any trials, it would render efficacy trials completely pointless.
In 2007, manufacturer Dendreon submitted powerful evidence attesting to the safety and efficacy of its immunotherapy drug Provenge, which targets prostate cancer. They were able to show that the drug resulted in a significant decline in deaths among its study population, which even persuaded the FDA advisory committee to weigh in on the application. But ultimately, the FDA rejected its application.
The FDA was unmoved by the evidence, simply because Dendreon didn’t properly specify beforehand what its study was trying to measure. Efficacy regulations state that finding a decline in deaths is not enough. The mountains of paperwork must be filled out just so and in the correct order. It took three more years and yet another large trial before the FDA finally approved the life-saving medication.
Due to all the additional costs imposed by the efficacy trial burden, Dendreon ultimately filed for chapter 11 bankruptcy.
In addition to the direct costs to companies, the extreme costs and financial risks imposed by efficacy trials have a huge chilling effect on investment in new drugs. If you’re an investment adviser trying to avoid losing your client’s retirement savings, you’re much better off investing in a more stable company like a bomb manufacturer building products to intentionally kill people than a drug developer trying to save lives. So it’s impossible to know all the treatments that never even got to an efficacy trial stage due to the effects of decreased investment due to the regulatory risks.
Conflicts of Interest
Long-term randomized trials are extremely expensive to set up and run. When billions of dollars in losses or gains are riding on the results of a study, this will almost inevitably influence the results. For example, an analysis of beverage studies, published in the journal PLOS Medicine, found that those funded by Coca-Cola, PepsiCo, the American Beverage Association, and the sugar industry were five times more likely to find no link between sugary drinks and weight gain than studies whose authors reported no financial conflicts.
The economic survival of the pharmaceutical company is dependent on the positive outcome of the trial. While there’s not a lot of evidence to support that there’s any illegal manipulation of results, it leads to two problems:
The Negative Results Black Hole
Here’s a fun fact that should be criminal:
- Negative trial results published: 37%
- Positive trial results published: 94%
- Money wasted repeating failed experiments: ~$100 billion annually
Companies are literally allowed to hide when drugs don’t work.
Company: “We tested this drug!”
FDA: “Did it work?”
Company: “…We tested this drug!”
This means we spend billions testing the same failed drugs over and over because nobody admits they failed. It’s like playing poker where everyone claims they won but nobody shows their cards.
Pharmaceutical companies bury negative results deeper than Jimmy Hoffa. So other companies waste billions testing the same dead ends.
Your insurance premiums are paying for this stupidity.
It’s like casinos only having to report when people win.
Countries That Don’t Have Our “Safety”
Japan’s Regenerative Medicine Act:
- Conditional approval after Phase II
- Real-world data collection required
- Time to patient: 2-3 years
- Americans fly there for treatment
- Terminal patients can access experimental drugs
- Doctor’s discretion, not bureaucrat’s
- Thousands of lives extended
- FDA: “But what if they die?” (They’re already dying)
- Passed despite FDA opposition
- FDA response: Made it effectively impossible to use
- Patients helped: <200 total
- Patients who wanted help: Tens of thousands
The COVID Test Fiasco: A Case Study in Excellence
Timeline of FDA’s COVID response:
- January 2020: WHO develops test, world starts using it
- February 2020: FDA blocks all non-CDC tests
- Late February: CDC tests contaminated, don’t work
- March: Private labs begging to test, FDA says no
- Late March: FDA finally allows other tests
- Cost: Unknown thousands of preventable deaths
The FDA’s defense: “We were ensuring quality.” The CDC’s tests literally didn’t work. You can’t ensure quality of nothing.
Solutions That Would Work (But Never Will)
Reciprocal Approval: If EU/UK/Japan/Canada approves it, we approve it
- Lives saved: Thousands annually
- FDA response: “But their standards might be 0.01% different!”
Provisional Approval: Let dying people try experimental drugs
- Current system: Die safely
- Proposed system: Maybe die, maybe live
- FDA logic: Death is preferable to uncertainty
Outcome-Based Approval: If it works in real patients, approve it
- Current: Does it work in cherry-picked trial patients?
- Better: Does it work in actual sick humans?
- FDA: “That’s not how we’ve always done it”
The Bottom Line: Safety Theater
The FDA doesn’t make drugs safe. It makes them:
- Expensive ($2.6 billion)
- Slow (9+ years approval phase, 17 years total)
- Inaccessible (95% of rare diseases have zero treatments)
- Ineffective (tested on wrong populations)
It’s security theater but for medicine. The TSA of healthcare. Except when TSA fails, your flight is delayed. When FDA fails, you die.
But at least the bureaucrats are safe from blame. And isn’t that what really matters?
P.S. - The FDA will likely object to this chapter. Estimated time for their formal response: 12-15 years, pending Phase III review.